|
|
Journal Home Contents Preview Next |
Pro Otology
Balkan Journal of Otology & Neuro-Otology, Vol. 3, No 1:18-20 © 2003
All rights reserved. Published by Pro Otology Association
Molecular Genetic Aspects of Hereditary Hearing Impairment
*A. Koitschev, †P. Rouev, †A. Boteva, †P. Dimov, *M. Pfister
*Department of Otorhinolaryngology, Head and Neck Surgery, University of Tuebingen, Tuebingen, Germany
†Clinic of Otorhinolaryngology, University Hospital, Stara Zagora, Bulgaria
ABSTRACT
Objective: The following article is a brief overview on diagnostic considerations in regard of patients with probable hereditary hearing impairment.
Data sources: An international team works since 1996 on establishing of broad database of families affected by HI. The data are collected according to standardised guidelines. This approach is determined to allow a reliable linkage of phenotype and genotype and lead to new aspects of molecular diagnostic in Hearing impairment (HI). The literature used is from the last 11 years.
Study selection: The review comprised the information from printed articles as well as online database.
Conclusions: Causal treatment options can only be expected by improving the scientific basis at the molecular level. The further development of a phenotype database will allow the collection of epidemiological data for genetic studies and phenotype/genotype correlation.The achievement of this data is the basic and most important tool in the identification and characterization of new genetic risk factors, e.g., hearing loss genes and their loci.
Key words: Hearing impairment (HI), Syndromic and Nonsyndromic HI, Hearing loss loci.
Pro Otology 1: 18-20, 2003
INTRODUCTION
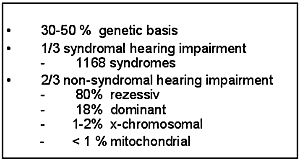
| Table 1. Congenital hearing impairment. |
|
Hereditary hearing impairment is a clinically and genetically heterogeneous entity and it is thought that at least one third of all hearing impairment in humans has a genetic basis (1). About 1 to 2 in 1000 newborn children are estimated to have a prelingual severe to profound hereditary hearing impairment (Table 1) (2).
Clinically, syndromic and non-syndromic hereditary hearing impairment can be differentiated. Up to 33% of all hereditary hearing impairment is syndromic. This entity includes 1168 syndromes with otologic manifestations (Online London Dysmorphology Database). The remaining 67% represents non-syndromic hearing impairment with no additional clinical features besides hearing impairment.
Non-syndromic hearing impairment is further classified by the mode of inheritance in autosomal-dominant (18%), autosomal-recessive (80%), X-linked (1-2%) and mitochondrial (<1%) hearing impairment.
A good overview on syndromic and non-syndromic hearing impairment loci is accessible through the Hereditary Hearing Loss Homepage.
Molecular Genetics of Hereditary Hearing Impairment
In syndromic hearing impairment more than 100 genes have been identified since 1990, showing a large heterogeneity even in the same type of syndromic hearing impairment, e.g. Usher-Syndrome type I a-f with 6 different genetic loci. However, the molecular genetic basis of most syndromes is still unknown.
|
|
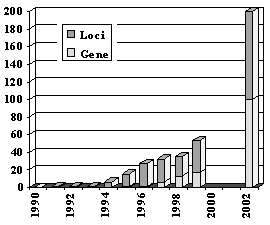
FIG 1. Development of the Identification of hearing loss genes. |
In non-syndromic hearing impairment, 56 autosomal dominant, 39 autosomal recessive, 5 X-linked loci and 2 mitochondrial loci have been mapped on the human genome. In total, 29 non-syndromic genes have been identified since 1994. All-in, more than 150 functional important genes of the inner ear are expected (FIG. 1).
So far the identified functional important genes (3) of the inner ear can be categorized according to their presumed function as:
a) Genes encoding for structural proteins: DIAPH, TECTA, COCH, COL11A1, COL11A2, COL2A1, COL4A3, COL4A4, COL4A5, NDP, USH2A, OTOG, OTOA, DSPP, USH1C
b) Motor molecule genes: MYO7A, MYO15, MYH9, MYO6, MYO3A
c) Genes involved in ion exchange processes: GJA1, GJB1, GJB2, GJB3, GJB6, KCNQ4, KCNQ1, HERG, SCN5A, KCNE1, KCNE2, CLDN14, SLC26A4 and ATP6B1
d) Genes encoding for transcription factors and factors for regulation of development: POU3F4, POU4F3, EYA1, EYA4, PAX3, MITF, SOX10, EDNRB, EDN3, FGFR3 and TFCP2L3
e) Unknown Function: CDH23, PCDH15, DFNA5, OTOF, STRC, TCOF, TIMM8A, TMC1, TMPRSS3, USH3A, WFS1 and TMIE
f) Genes encoded by the mitochondrial genome: tRNALeu, tRNALys, tRNASer, tRNAGlu and 12SrRNA
g) Hearing impairment genes in cooperation with modifier genes: DFNB26 and DFNM1
Mutations in one particular gene, connexin 26, have been shown as a major cause of inherited and sporadic non-syndromic hearing impairment in different populations (4). The CX26 gene encodes the gap junction protein connexin 26 (beta-2, GJB2) whose expression was shown in several tissues and in the cochlea. One particular mutation, 35delG, is the most frequent mutation in the CX26 gene. It represents a deletion of a guanosine (G) in a sequence of six G extending from position 30 to 35 of the CX26 cDNA. The deletion creates a frame shift resulting in a premature stop codon and a non-functional intracellular domain in the protein. The 35delG mutation can be detected at the molecular level using PCR followed by sequencing or other reported diagnostic procedures (see molecular diagnostics). Due to current data from the US and Europe the frequency of this mutation varies in geographically distinct populations.
Clinically, homozygous patients with the 35delG mutation show a variable phenotype, ranging from mild to profound hearing impairment. However, most patients with homozygous 35delG mutation show a severe to profound hearing loss phenotype.
Regarding other dominant and recessive hearing loss loci, there is an overlap and it has been shown that at least in 3 cases (MYO7A, GJB2, TECTA) the same gene but a different mutation can lead to a recessive or dominant hearing impairment. For this reason, a hearing loss gene can not necessarily be associated with a phenotype.
Phenotype in hereditary hearing impairment
The clinical classification of non syndromic hearing impairment takes the following criteria into account: Severity of hearing impairment, age of onset, type of HI, frequencies involved, unilateral/bilateral, stable/progressive, syndromic/non-syndromic (5, 6).
In general, recessive HI tends to be more severe than dominant HI. This might be due to the fact that in recessive cases both alleles harbor the disease causing mutation in comparison to only one allele in dominant HI.
Moreover recessive HI is characterized predominantly by a congenital or prelingual onset versus dominant HI with pre- and more often postlingual onset.
In X-linked HI, sex difference in HI can be detected due to the fact that affected males are homozygous and affected females are heterozygous for the disease causing allele. For this reason affected females show a less severe or even mild phenotype in comparison to affected males, who carry only the disease causing allele.
Prognostic statements on the progression of the disease are locus and inheritance dependent. In general, recessive hearing impairment tends to be stable versus dominant hearing impairment with a progressive pattern. So far, only families with mutations in TECTA show a non-progressive type of autosomal dominant hearing impairment.
Radiological assessment
Computer tomography scans (CT) are valuable in revealing Mondini’s deafness, Michel deafness and bulbous inner auditory canal or absent cribrose areas in the cochlea. In particular, x-linked hearing impairment (DFN3) has characteristic features including a bulbous inner auditory canal as well as a small cochlea. In case of a DFN3 phenotype a stapedectomy is a contraindication due to the reported perilymphatic gusher.
In case of large vestibular aqueduct, an association between this phenotype and mutations in the PDS gene has been established.
Molecular Diagnostics
All identified hearing loss loci can be analyzed and risk calculations can be performed in families with hereditary hearing impairment in which significant cosegregation to a hearing loss locus was established. A major limitation of this analysis is often the size of a family. For this reason a reference centre should be contacted before such an analysis is intended.
Regarding all identified functional important genes of the inner ear, mutation analysis is in principle possible.
However in daily practice, only mutation analysis of Connexin 26 is currently suitable due to the epidemiological importance of this gene as well the established low cost screening techniques (e.g. SSCP, restriction test, sequencing).
DFNB1, the hearing loss locus for Connexin 26 as hearing loss gene, accounts for 50 to 80% of all autosomal recessive hearing impairment. Up to 70% of these cases are due to one single mutation, a deletion of a single guanine nucleotide in position 30 to 35, called 35delG. In addition, up to 10 % of sporadic cases of severe to profound hearing impairment harbors the 30delG mutation in both alleles (7).
Therefore this test is the first genetic test clinically available for children affected by sporadic non syndromic hearing impairment.
Outlook
In industrialized countries nearly 20% of the population suffers from hearing loss. Due to a lack of knowledge about the molecular basis of hearing loss, our present diagnostic and therapeutic tools are limited and make hearing impairment one of the most poorly treatable human health problems. With already a fifth of the total population affected and an expected increase as a result of current demographic and lifestyle changes, the severe impact on social and professional life will make hearing loss the major health concern of the communication age. This will particularly affect the social integration of the ageing population in the future. Causal treatment options can only be expected by improving the scientific basis at the molecular level.
Consequently, the further development of a phenotype database will allow the collection of epidemiological data for genetic studies and phenotype/genotype correlation. The achievement of this data is the basic and most important tool in the identification and characterization of new genetic risk factors, e.g., hearing loss genes and their loci.
REFERENCES
Cohen M, Gorlin R. Epidemiology, Aetiology and Genetic Pattern. In: Gorlin R, Toriello H, Cohen M, eds. Hereditary Hearing Loss and its Syndromes. Oxford: Oxford University Press, 1995:9-21.
Reardon W. Genetic deafness. Journal of Medical Genetics 1992;29:521-6.
Haack B, Pfister M, Blin N, Kupka S. Genes involved in hereditary hearing impairment. Current Genomics 2003; 4(5).
Estivill X, Fortina P, Surrey S, et al. Connexin 26 mutations in sporadic and inherited sensorineural deafness. Lancet 1998; 351:394-8.
Calzolari E. European Work Group on the Genetics of Hearing Impairment. Group 4a: study group on craniofacial malformations and hearing impairment. European Work Group on the Genetics of Hearing Impairment Infoletter 1997;3:1-28
Grundfast KM, Lalwani AK. Practical Approach to Diagnosis and Management of Hereditary Hearing Impairment (HHI). Ear Nose and Throat Journal, 1992;71(10):306-18.
Kupka S, Braun S, Aberle S, et al. Frequencies of GJB2 mutations in German control individuals and patients showing sporadic non-syndromic hearing impairment. Hum Mutat 2002;20:77-78.
|
Pro Otology |
Journal Home Contents Preview Next |